
If there’s an option to write a review or to blog, after reviewing the same number of published articles, I will most definitely choose to blog.
Why?
Because of the freedom to voice my opinions. Also, I can start a sentence with “because”, even if it is grammatically incorrect. Review writing is also a process of accumulating all the information you have read for your project and providing an unbiased conclusion based on evidence. I plan to be unbiased here but not everything has to go according to “plan.” Science does not. Thus I shall review from personal experience as well. After all, that is the novelty of blogging.
***
Hello, dear reader. Hope you are persevering!
Today I would like to share my experience using bioinformatics tools.
One cannot start an article on bioinformatics without mention of Dr. Margaret Oakley Dayhoff (1925-1983), the lady who pioneered the field of using computational methods to the field of biochemistry. Although she began by using computers in chemistry, she, along with physicist Robert S. Ledley, brought these resources against biomedical problems [1,2].
The first project, COMPROTEIN, was developed to determine the primary structure of protein. The software also used three-letter codes for proteins (later Dayhoff simplified the data by using one-letter codes to create the first ever Atlas of Protein Sequence and Structure by Dayhoff and Eck in 1965) [3].
Apart from the fact that she was a woman in software (don’t be shocked, there are ladies making millions in software in 2023. Check out this Forbes list), there are actually some really interesting tools to study biological data on your computers! I’m just glad it was a lady who put it in motion [just like Dr. Rosalind Franklin for Watson and Crick’s structure of the DNA helix].
***
I believe I am dexterous and a hard worker, but I have never been able to perform complete projects in wet lab. When I began my scientific journey, the lock down caused most undergraduate colleges in India to revert to bioinformatics tools. My project work took an in-silico approach. Most of my knowledge in wet lab facilities is from regular class lab-work. Time flies when I’m in the lab. Weirdly, disinfecting lab benches is something I miss.
During my masters, I continued in-silico for my project, even though my actual degree- molecular biology and human genetics, provided me ample experience in wet-lab.
This experience has provided me an all-round expertise in when and how to use wet-lab or dry-lab. While confirmatory studies can only be performed in wet-lab, proficiently using computational tools can provide some promising preliminary studies. The extensive information accumulated can be used to formulate a study plan for wet-lab studies, for a conservative and judicious use of resources for hypothesis testing.
***
Bioinformatics approaches along with tools that I found fascinating-
1. Curating and categorizing sequence information onto databases
Three massive online databases that store information about various genes, proteins, genome assemblies, RNA, expression data are National Center for Biotechnology Information (NCBI, but mainly GenBank), European Molecular Biology Laboratory (EMBL) and DNA Data Bank of Japan (DDBJ).
Most information uploaded on any of these databases is synced daily for a collaborated effort in the International Nucleotide Sequence Database Collaboration (although this is mainly for nucleotide sequence data).
Personally, I collect data from NCBI sources and use the tools provided by EMBL which by far, are outstanding.
I’m sure most of you understand how easy cloud storage has made data syncing into all your devices (Apple and Google users, you know). Similarly, online storage of biological data also aided in the Human Genome Project (HGP). The human genome consists of approximately 3.3 billion bases (~3GB of data) and it was the collaborative effort of various labs across the globe! Sequenced data from one lab could be easily accessed by another lab halfway across the world because of online databases where information could be stored! (Side story: The first (incomplete) human genome was mapped in 2001. Last year (2022), scientists have finally deducted the complete sequence!)
This has honestly been a huge help, especially in my project wherein I needed to analyze the expression studies conducted across various demographic groups. The expression data helped in understanding the essential or conserved sequences how some mutations are beneficial for bacterial survival or virulence.
2. To study evolution
Evolution is studied by analyzing something called homology which is to find out the similarity. The more similar two things, the closer they are through evolution (how scientists figured chimps and humans were related) or conserved regions (sequences used to predict certain organisms, like how one knows Neisseria gonorrhoeae and Neisseria meningitidis both belong to genus Neisseria). MEGAx is a software that can develop evolutionary (phylogenetic) trees. For example, depending on the IgG antibody, the following depicts the homology between the African elephant, orcas, mice, humans, chimpanzees, lions and leopards-
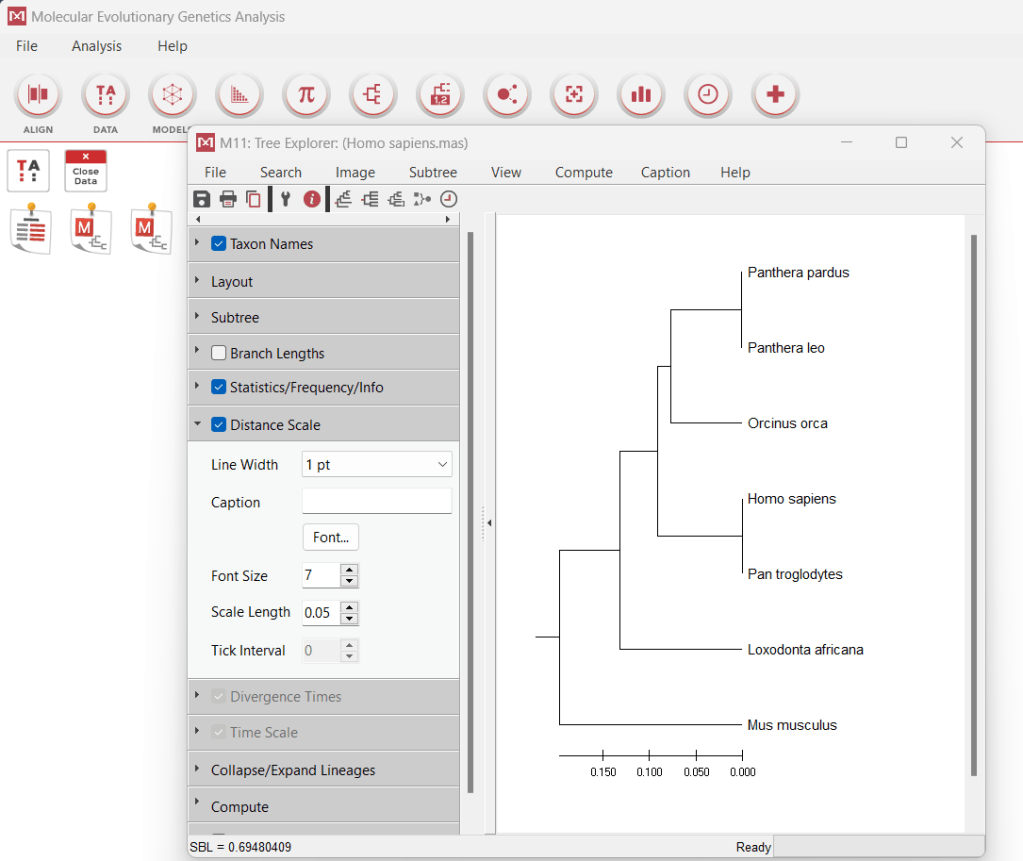
This has been used to predict the effect of mutations. In the case of SARS-CoV-2, particular mutations (quite literally single-digit) produced a severe viral strain that could spread the infection faster but another mutation could also make it less fatal. All this depends on pattern-based algorithms but more on that in the next part.
3. Protein structure prediction
Honestly, I find this tool most fascinating. I have worked with two softwares- PyMol and Discovery Studio Visualizer (DSV). While RCSB PDB (Protein Data Bank) is an online platform that provides structural information and one can even download the file to use for PyMol. For example, here’s an example of a beta-2 adrenergic receptor (AR) and it’s interaction with propranolol (beta-blocker). The fluoroscent green spiraling structure (ribbon) is the receptor.

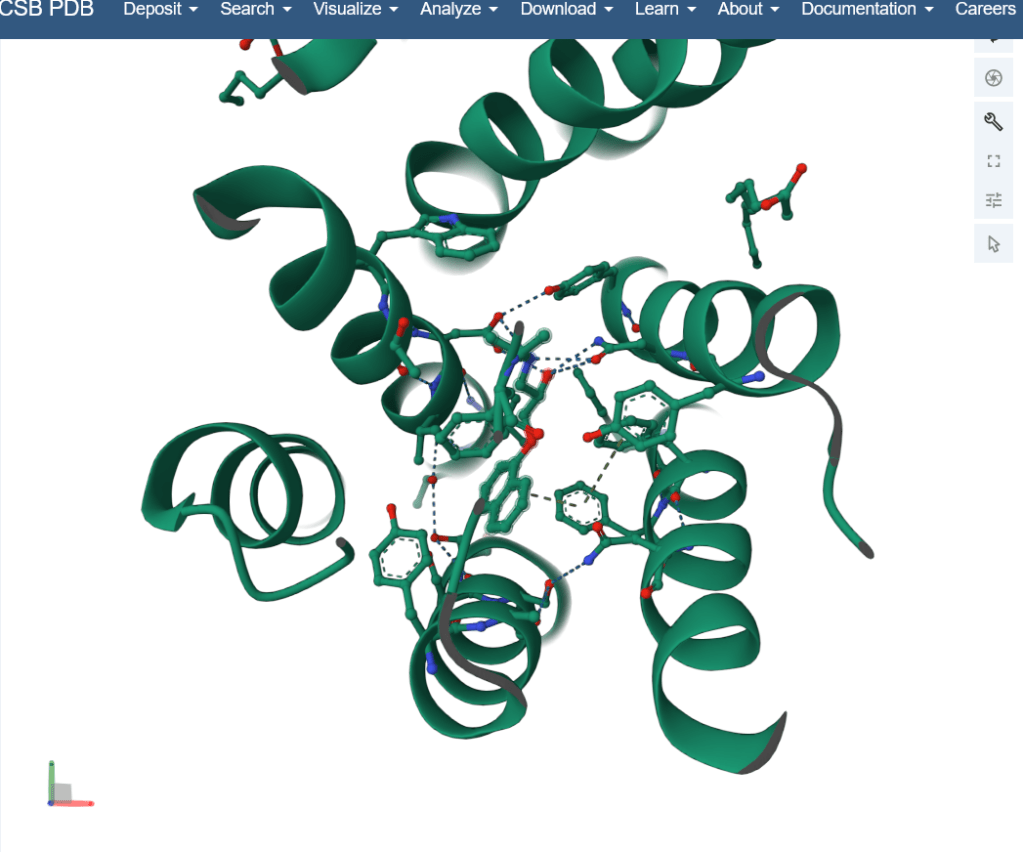
Structural prediction allows one to visualize how certain drugs interact with the cells in our body. While most prediction tools are based on previously collected data and homology (similarity) between sequences, finding similar structures can also predict the function of the protein.

Structural bioinformatics has been used to hypothesize drug targets and drug interactions. It’s use in focusing on feasible targets with least adverse effects can be extremely helpful for pharmaceuticals. One of the best websites for diagnostics and therapeutics is OpenTargets.
4. Performing (predicting) certain wet-lab experiments
Lastly, performing certain wet-lab tasks in-silico such as NCBI’s primer designing and PCR reactions. This method has made it easier to target a gene and analyze the progress of PCR in seconds instead of waiting for two hours for results.
You can even perform a restriction digestion for a particular plasmid to find out how the bands should look. For example, the latest version of the NEB cutter 3.0 looks super sleek and easy to use. I tried to see the effect of REs (Bam H1 and AhdI) on pUC19 and this is a virtual digestion and image if run on 1% agarose gel (and it took me 5 seconds to build)-

Something I really like playing around with are the effects of mutations on the protein structure. The Swiss Institute of Bioinformatics (SIB) provides many tools for protein studies. One of them is the SWISS-MODEL that predicts protein structure.
Finally, here’s an overview of some other applications in the field of bioinformatics-
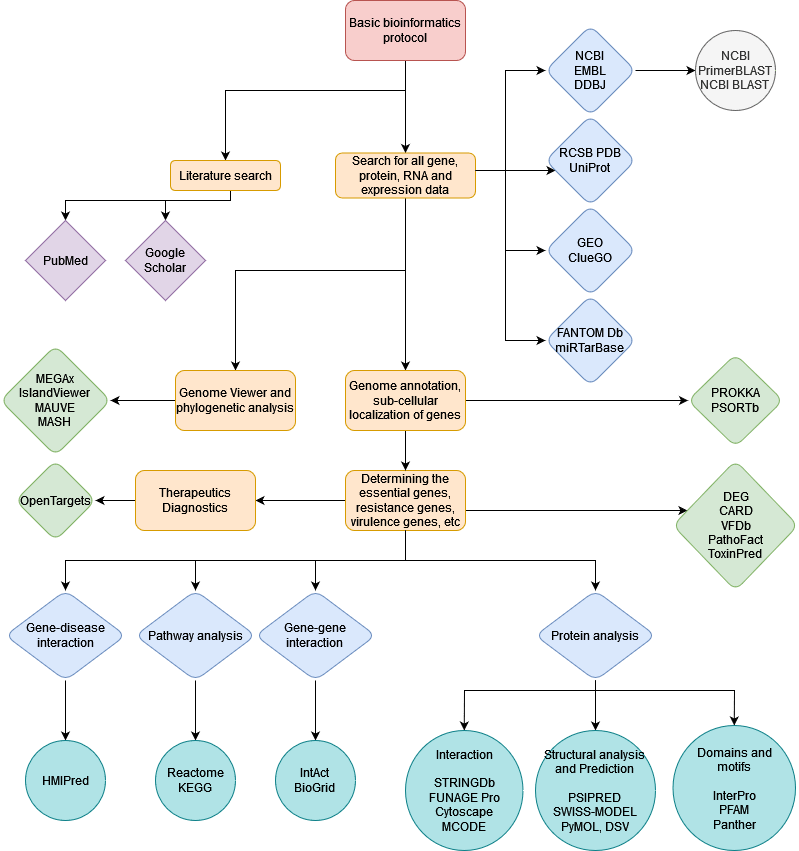
***
Future of bioinformatics- systems biology
Systems biology is used to describe a holistic or “cross-disciplinary” approach for biology. The approach was initially used to integrate vast amount of “omic” (genome, transcriptome/RNA, proteome) data and determine it’s function. In short, dealing the complex biological data, top-to-bottom or bottom-to-top[4]. Weirdly, as a molecular biologist, this approach is probably the exact opposite of my field.
Mathematical modelling is being used to study or represent biological data. For example, making metabolic networks or signaling pathways, something like the network below for breast cancer from KEGG-
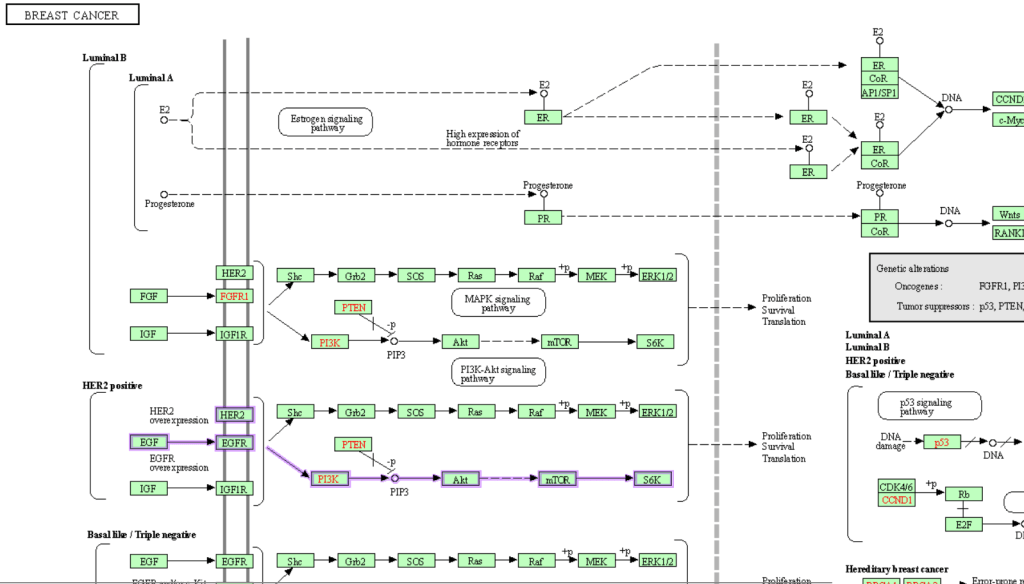
Till now, every aspect of biology has been stored in online databases separately. One wants to study proteins, go to UniProt, pathways, to Reactome. Systems biology is a way to combine all these components using computational and mathematical tools to create “complex dynamic networks”. In a way, to study the interactions between different molecules in the cell- genes, transcripts, RNA, proteins and metabolites from a molecular perspective and to create better models which can decipher the phenotypic implications of these interactions[4].
***
While I am a budding scientist who prefers wet-lab work, however tedious and time-consuming, I cannot deny that dry-lab has been developed for our convenience- to alleviate some of the workload. As a scientific community, instead of shunning it or not giving it enough credit, we should learn these new skills to boost our productivity and inculcate them into practice.
Just because one is a biologist who cannot code, doesn’t mean one cannot learn to or take help from another who does. Research is moving towards an interdisciplinary approach and integrating these novel approaches may actually aid the entire community for faster output.
***
REFERENCES:
1. Oakley, M. B., & Kimball, G. E. (1949). Punched card calculation of resonance energies. The Journal of Chemical Physics, 17(8), 706-717.
2. Ledley, R. S. (1959). Digital Electronic Computers in Biomedical Science: Computers make solutions to complex biomedical problems feasible, but obstacles curb widespread use. Science, 130(3384), 1225-1234.
3. Gauthier, J., Vincent, A. T., Charette, S. J., & Derome, N. (2019). A brief history of bioinformatics. Briefings in bioinformatics, 20(6), 1981-1996.
4. Likić, V. A., McConville, M. J., Lithgow, T., & Bacic, A. (2010). Systems biology: the next frontier for bioinformatics. Advances in bioinformatics, 2010.


Leave a comment